Linck Lab
Montana State University ethanblinck@gmail.com
Inferring population structure – the subdivision of a species into groups of individuals
interbreeding with each other at a higher frequency than expected by chance – is a fundamental
goal of population genetics. Beyond its obvious relevance to systematics, taxonomy, and conservation,
understanding patterns of population structure is crucial for a range of applications, from
detecting selection and migration to identifying genetic variants associated with specific traits in
genome-wide association studies (the red-hot “GWAS” trend).
While on the one hand, the wealth of data resulting from the advent of next generation sequencing
technology has proved a boon for describing subdivision in nonmodel organisms, it has also
provided challenges. For example, many computational methods for detecting structure were developed prior to the genomics
era and may not be suited to the unique characteristics of large SNP datasets. Of these,
Pritchard et al.’s structure is the most widely cited,
and the basis for a raft of other methods that feature an underlying generative model and explicitly
estimate a suite of population genetic parameters.
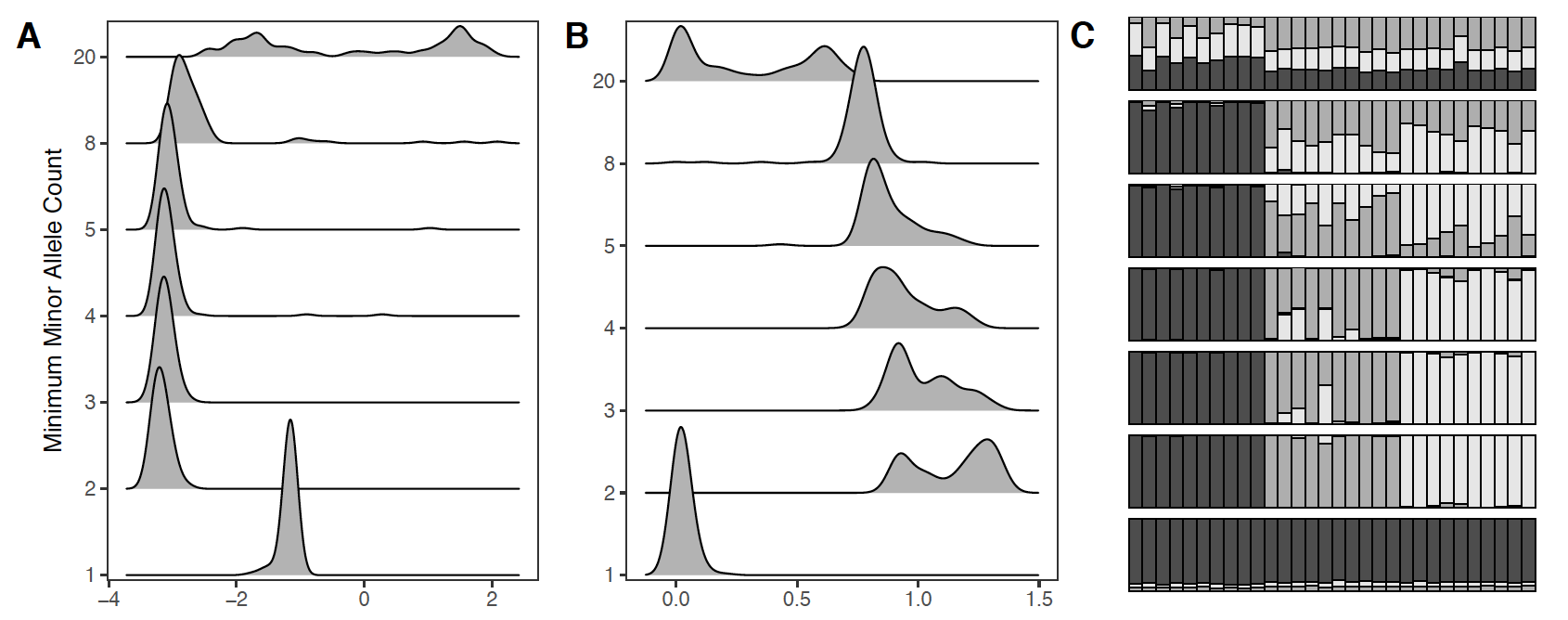 Figure 2 from our preprint, showing inferred (A) population discrimination and (B) admixture across different MAF levels.
Figure 2 from our preprint, showing inferred (A) population discrimination and (B) admixture across different MAF levels.
Unfortunately, at least in our lab’s experience, these model-based approaches frequently fail with modern datasets produced by methods like RADseq. In particular, we’ve observed a pattern where all individuals have the great majority of their ancestry assigned to a single population, producing a “smear” pattern with the remaining small percentage assigned to informative clustering results. Minor allele frequency cutoffs (or selecting parsimony informative sites alone) appeared to partially help – perhaps reflecting theoretical predictions that different frequency classes of alleles reflect different evolutionary histories and levels of subdivision. But we lacked a real rationale for applying these filters while processing our data, and lacked an understanding of what different levels of filtering actually did.
So to address these issues, C.J. and I used both simulated
and empirical (Golden-crowned Kinglets!)
SNP datasets to explore how MAF choice affected our ability to recover
known population limits. We looked at both programs like structure and non-model-based approaches
like k-means clustering with principal components. We found the former class of methods are indeed very
sensitive, but the latter class are fairly robust, speculated on why, and made a few recommendations
for best practices. You can read our new preprint here –
we’re hoping to submit it soon, but would love feedback in the mean time.
{kind=link}